Research Projects
Some examples of the past and present projects of our group, intended to give you a sense of the types of work we do and hopefully whet your curiosity. Of course, you can find many more details in our publications.
Table of Contents
Polymer Nanocomposites for Ballistic Impact Resistance
Polyurethanes are commonly used in transparent armor systems to increase the survivability, blast resistance, multi-hit resistance and fragments containment of multi-layered systems. The high tensile-ductility, fracture toughness and self-healing ability are key-factors that determine the performance of polyurethanes under high-strain rate conditions. Polyurethanes are block copolymers formed by hard and soft segments, respectively comprised of diisocyanate-chain extender and polyol moieties. The resultant domain-segmented macromolecular architecture will depend on many factors, such as: degree of phase separation, composition and relative content of hard and soft segments and synthesis conditions. Our research focuses on understanding the influence of macromolecular structural features in the mechanical behaviour of polyurethane under dynamic loading conditions. Different compositions of polyurethanes based on aromatic and aliphatic diisocyanates are being synthesized and tested via ballistic and spall testing in order to establish a microstructural/performance relation. For instance, we’ve studied the ability of Halloysite nanoparticles to increase the survival of polyurethane under spall fracture through extensive micro-crack formation and prevention of crack coalescence by nanotubes crack-bridging. We’ve also looked at the role of a low concentration of Halloysite nanotubes in determining the resultant spherulitic structure of partially silane-terminated polyurethanes, and the subsequent effects on the spall strength and fracture toughness under strain rate magnitudes of 104 s-1.




The project is sponsored by NSERC’s “Discovery Grant” program.
[1] Aguiar, R., Lebar, A., Oddy, A., Miller, R. E. & Petel, O. E. Synthesis and Mechanical Characterization of Polyurethane Reinforced with Halloysite Nanotubes. AIP Conference Proceedings 2272 (1), 120001 (2020).
[2] Aguiar, R., Miller, R. E. & Petel, O. E. Synthesis and Characterization of Partially Silane-Terminated Polyurethanes Reinforced with Acid-Treated Halloysite Nanotubes for Transparent Armour Systems. Sci. Rep. 10, 13805 (2020).
Atomistic Modelling of Dynamic Failure in Polymers and Polymer/Metal Multilayers
Spall of amorphous polymers
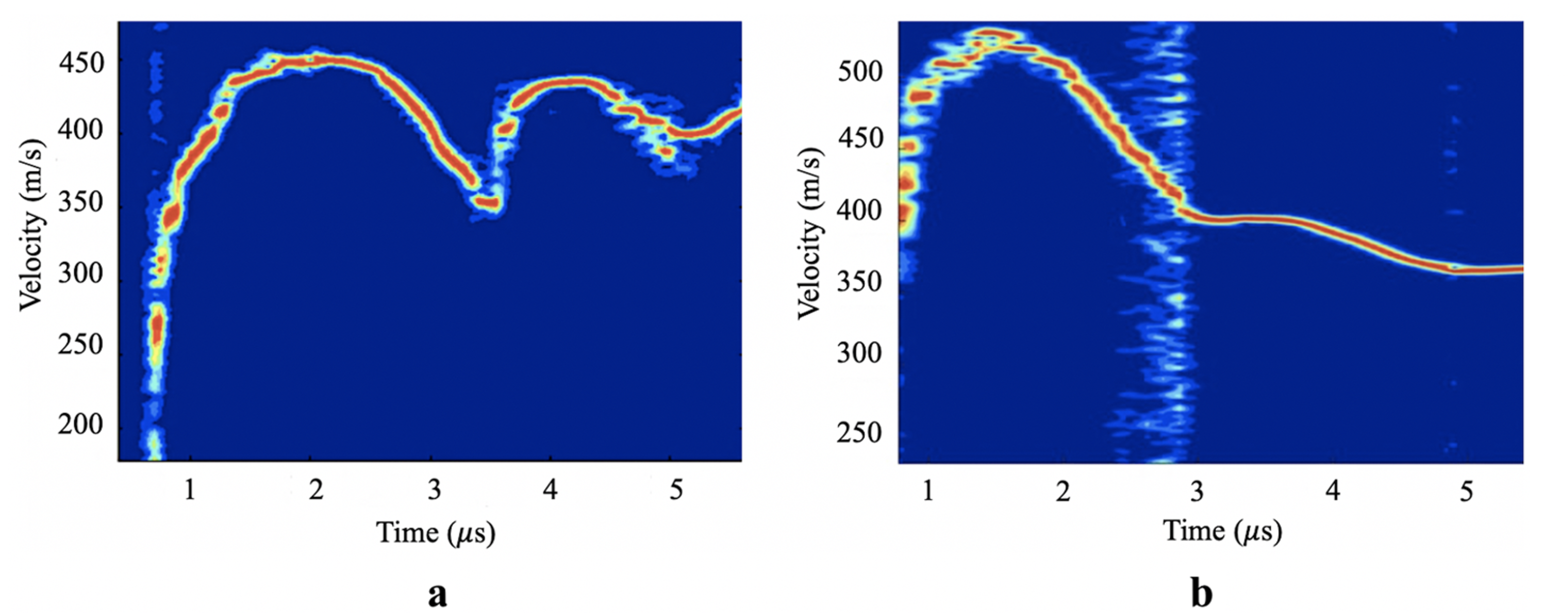
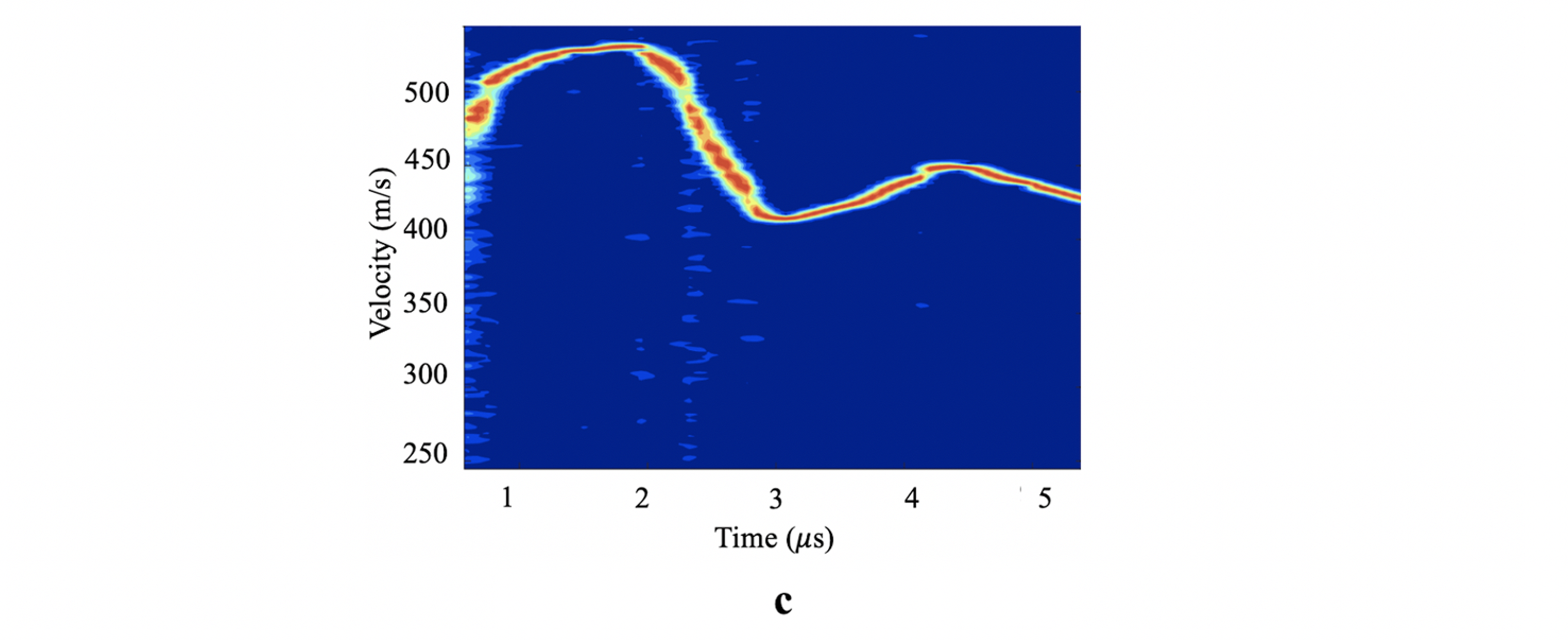
A gas gun experiment uses impact between a flyer and target plate to induce high strain-rate failure through spall in the target. We used MD simulations of the same process at the nanoscale to explore how amorphous polymers fail at high strain-rate.
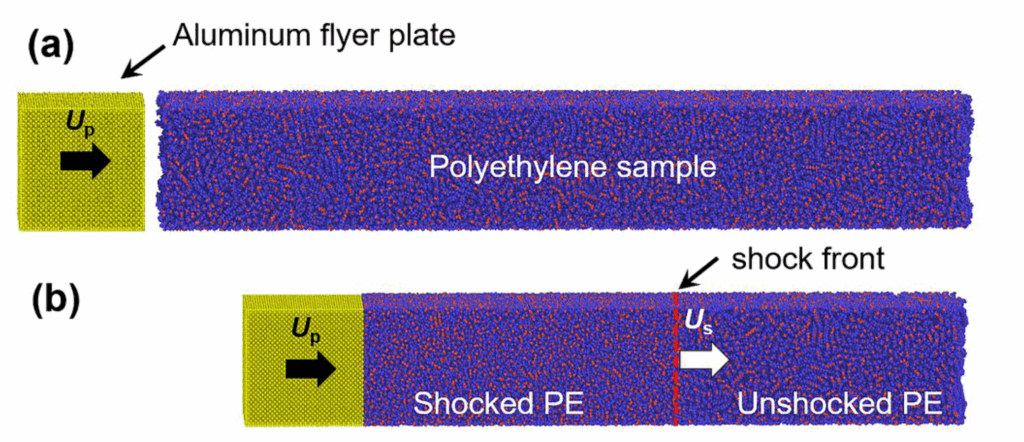
Our simulations allowed us to probe a basic assumption in shock experiments: that the spall stress can be estimated from the free surface velocity (which is really the only thing you can measure during these high speed experiments). An example from our simulation is shown below.
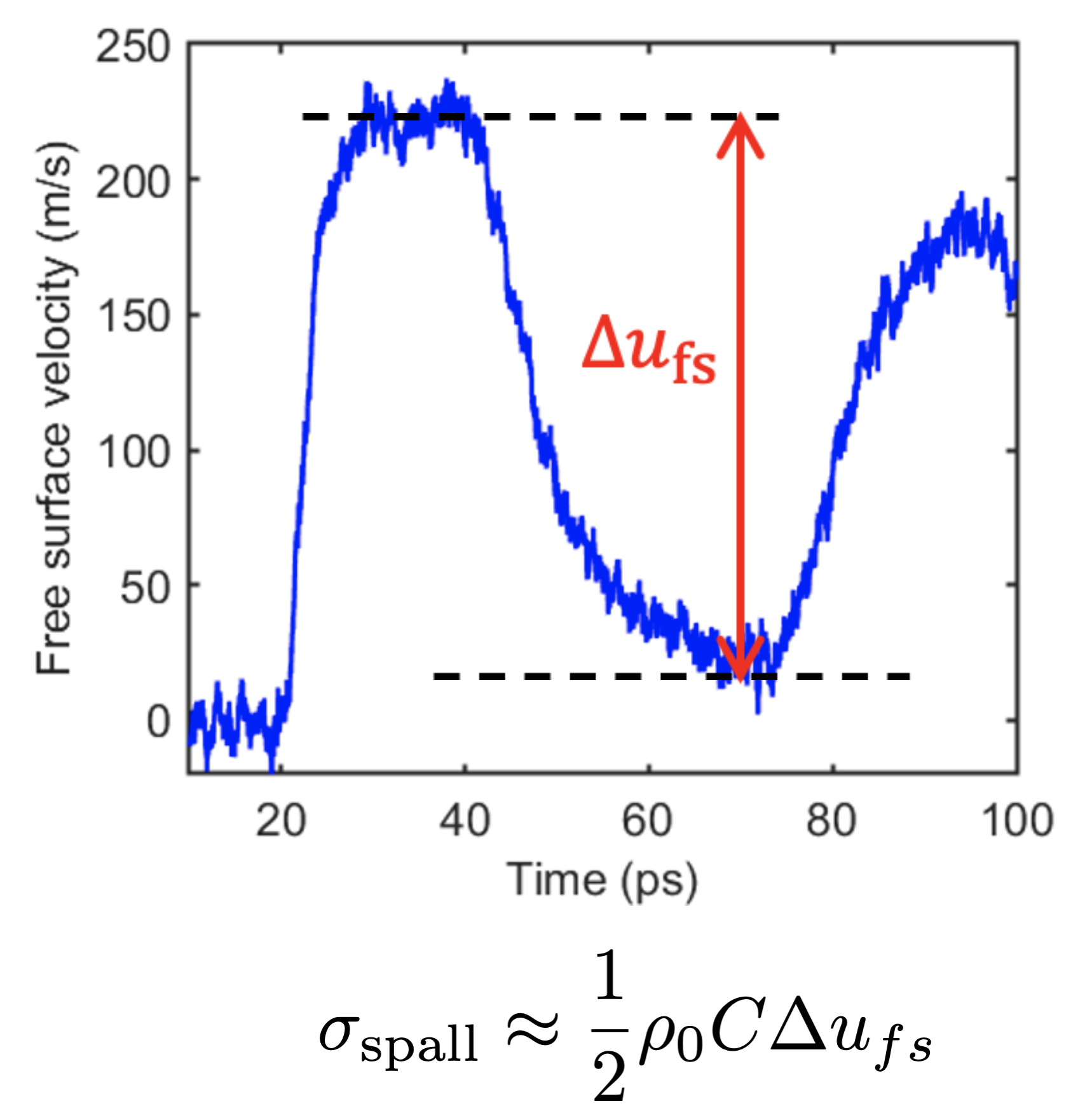
But MD simulation gives you access to much more information than the experiment, allowing us to compute the stresses directly. We find that the free surface velocity estimate can be substantially different from the observed stresses depending on the failure mechanism of the material. This difference can be especially significant in polymers because of their structure and failure mechanisms. Below, “direct” and “highest” are from the MD data, while “indirect” comes from the free surface velocity.
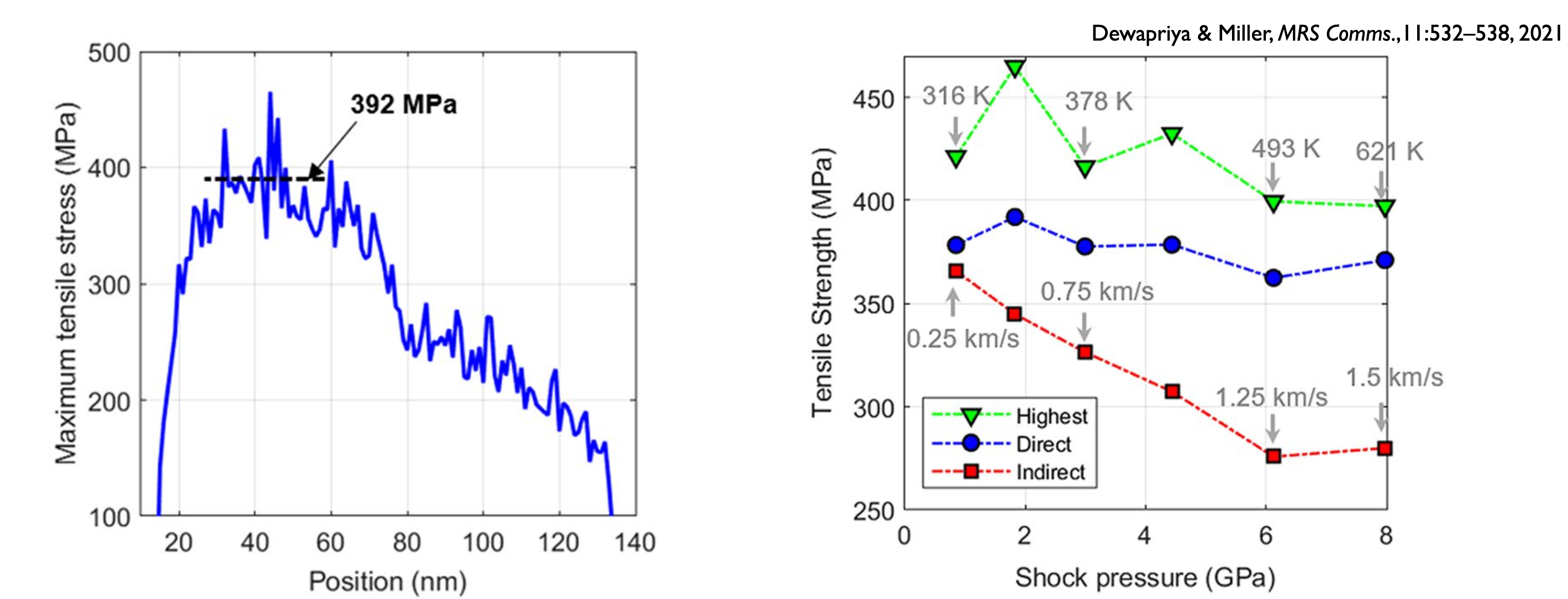

The project is sponsored, in part, by NSERC’s “Discovery Grant” program.
Mechanics of nanoscale multilayered structures under high strain-rates
Material’s ability to withstand high strain-rate loading is essential for numerous applications ranging from protecting consumer electronics against accidental shocks (e.g., dropping a mobile phone) to shielding space structures against the hypervelocity impacts of micrometeorites. The benefits of conventional protective materials such as Kevlar and ceramics have been extensively exploited. Recent advances in impact testing at small scales have led researchers to explore the dynamic mechanical behavior of nanomaterials facilitating the development of the next generation of armor materials. For example, microprojectile impact tests of ultrathin polymer films have revealed that their specific penetration energy is about ten times more than that of the conventional armor materials. On the other hand, metallic nanostructures possess extraordinary mechanical properties. Therefore, structures with superior ballistic performance could be achieved by multilayer arrangements of nanoscale polymer and metal films. In order to test this hypothesis, we simulated the impact tests of nanoscopic polymer/metal multilayers as well as polymer/ceramic multilayers using molecular dynamics.
Our simulations demonstrated that the ballistic limit velocity and the specific penetration energy of the multilayer polymer/metal nanostructures are significantly higher than the experimentally measured values for any material. The findings have been published in the Journal of Applied Mechanics. The video abstract of the paper is shown above.
Moreover, we found that an aluminum layer confined between two polyurea layers has approximately 40% less impact-induced dislocations compared to an aluminum layer which is in contact with only one polyurea layer. This observation clearly demonstrates that the polymer exerts remarkable resistance on the dislocation nucleation in the metallic layer. The findings have been published in Computational Materials Science. The video abstract of the paper is shown below.
This project showed us that modeling material interfaces could be challenging even for someone with a background in atomic simulations. Therefore, we shared some useful information and the LAMMPS input files, on GitHub, to model the aluminum-polyurethane system shown in the video below.
The project is sponsored, in part, by NSERC’s “Discovery Grant” program.
Atomistic Modelling of Crystalline Metals
The role of hydrogen in fracture
Understanding and predicting fracture is, of course, of fundamental importance to engineering. The more accurate and reliable our understanding of the process, the better equipped we are to design lightweight, long-serving structures with confidence that they will not fail suddenly and catastrophically. Our research focusses on applying atomic scale models to advance our understanding of fundamental aspects of plasticity and fracture in structural engineering metals, specifically addressing the important role that hydrogen plays in making certain important metals more brittle. For example, we are working to address hydrogen embrittlement effects in aluminum alloys (so-called Hydrogen enhanced crack-tip activity or HECA) and the role of hydrides (precipitated hydrogen compounds) in the fracture behaviour of zirconium alloys. We use computer simulation techniques that have been developed in our group, as well as those developed by other researchers. For example, we use multiscale methods that extend the size of the simulation that can be run with atomistic accuracy. This is accomplished through judicious choice of which regions of the problem are treated using a fully atomistic description, allowing us to study hydrogen-aluminum interactions within realistic configurations of atoms around a stressed crack tip. As well, we employ computational methods to explore atomic configurations and accurately determine the activation energy associated with key processes like dislocation nucleation. Accurate knowledge of such data is of fundamental importance to any larger scale models aiming to predict fracture toughness. Finally, we are working to develop new, more accurate interatomic potentials to describe the zirconium-hydrogen system. This will allow us to study the complex interactions between cracks and embedded hydrides.
Project sponsored by NSERC’s “Discovery Grant” and “Discovery Accelerator Supplement” programs.
Nanotwinned Copper
If we ask the right questions and look at small or simplified systems, we can gain insight from purely atomistic studies of deformation. For example, we studied the effects of twin boundaries in so-called nanotwinned materials. These materials are of interest because they are very strong like their “nanocrystalline” brethren, but they are less brittle. The movie below is one example of such a simulation, but the real understanding comes from combining such visualizations with results such as the stress vs. strain curves as a function of twin spacing and orientation.
(video courtesy of Ishraq Shabib)

Project sponsored by NSERC’s “Discovery Grant” program and a PREA award from the government of Ontario.
Multiscale Modeling of Fracture and Failure in Crystalline Solids
Molecular dynamics (MD) and atomistic simulations have the potential to impact areas ranging from nanotechnology, to drug design, to the next generation of advanced materials. However, the predictive capabilities of MD are limited in several ways. The two most significant limitations are the prohibitively small numbers of atoms that can be studied and the limited availability (or sometimes complete lack of availability) of accurate interatomic potentials for many important combinations of atomic species. Our research pursues the development of methodologies, algorithms and tools that help to overcome these obstacles. At the same time, we use these tools to study important scientific questions related to the mechanical failure of materials at the nanoscale. These questions have consequences for the design of materials with enhanced properties, for our understanding of nanostructures, and for the fundamentals of fracture and failure in structural materials.
The Quasicontinuum (QC) Method
It is very difficult to study the failure of material from the perspective of atomic interactions because of the large number of atoms involved in any macroscopic deformation processes. Together with many collaborators, we have worked on the development of the quasicontinuum method. This is a “partitioned-domain, concurrent multiscale method” in the parlance of the field. Some regions of the problem are treated with a fully atomistic description and others use a more approximate model based on finite elements.
Below is an example of a QC simulation of fracture. At the start of the simulation, only the small region near the crack tip is modelled with full atomistic resolution. As the deformation proceeds, the model automatically adapts and grows the atomistic region to capture the bond breaking processes at the crack tip.
First, a broad view of the crack simulation:
And now a close-up of the crack tip. Notice how a dislocation is emitted from the crack tip every now and then (the shearing of the elements emanating from the crack tip).
See www.qcmethod.org for more details.
The Coupled Atomistic and Discrete Dislocation (CADD) method.
Like the QC method, CADD lets us study deformation and failure from an atomistic perspective at a reduced computational cost. CADD goes a step further than QC by allowing for two different models of a dislocation: either the fully atomistic description of the dislocation core or a simplified representation as a line defect in an elastic medium. CADD combines two approaches to multiscale mechanics: that of the QC method and that of the discrete dislocation modeling community. Our contributions to CADD were all 2D (see several of our contributions here, here, here and here). Since then, others have taken up the challenge of extending the model to 3D.
In the indentation example below, notice how the elements at the bottom of the simulation do not need to be refined to the atomic scale. Instead, dislocations are passing into the larger elements where they are treated as elastic defects.
Here is a view of just the atoms under the indenter:
Finally, here is a high-strain rate fracture simulation. There are several dislocations that can be seen leaving the crack tip, which are carried away as elastic defects thanks to the CADD formulation:
(video courtesy of Behrouz Shiari)
Project sponsored by NSERC’s “Discovery Grant” program.
Modeling the degradation of lubricating oils
Lubricating oils breakdown due to heat, shear stress, pressure and oxidation as they are squeezed between the moving parts of machinery. In effect, this is a long, slow chemical reaction that we would like to understand, in order to make predictive models of the process. On the one hand, such reactions occur at the level of molecular bond breaking, but on the other hand such reactions occur only rarely (compared to the time scales of atomic vibration) and in a myriad of possible ways.
We are trying to bring a number of different modeling techniques to bear on this problem. At the “macroscale” we are using chemical kinetics modeling to simulate the reaction process. However, such models require high-quality data (from either experiments or theory) to make their predictions accurate. As such, we are also using atomistic and DFT methods to compute key data such as activation energies, transition states, and minimum energy reaction paths.
Fluid mechanics at the molecular scale
We are using MD to try to understand the connection between composition of a fluid and its properties. The movie below is an example of an NEMD simulation of hexene that can be used to compute viscosity. The hexene-hexene interactions use the OPLS-aa potential, while a simple Buckingham potential is used to mimic the attraction between the fluid and the aluminum wall. Two hexene molecules are highlighted to better visualize their motion. See our paper on the subject for more details.
(video courtesy of Carlos Campana)
Project sponsored by NSERC’s “Engage” and “CRD” programs.
Finding transition states: The Transition Rapidly-exploring Random Eigenvector Assisted Tree (TRREAT) method
Although the name of the method is a bit of a mouthful, we couldn’t resist the resulting acronym. Inspired by other eigenvector following methods and the previously developed TRRT algorithm, TRREAT is a method for efficiently exploring molecular conformations and reaction paths to find previously unknown transition states, reaction products and important quantities like activation energies.
The method explores the configuration space aggressively but intelligently, and produces reaction pathways that serve as approximations to the minimum energy pathway. The TRREAT paths can be used as high quality initial guesses for methods like the nudged elastic band (NEB) method, so that the combination of TRREAT and NEB can be used to obtain excellent MEP results.
The TRREAT method was recently published here.
Project sponsored by the Ontario Centres of Excellence and NSERC’s “CRD” program.